FDA recall requirements (e.g., medications, devices, supplies, supplements, classifications)
The U.S. Food and Drug Administration (FDA) oversees recalls for various products, including medications, medical devices, medical supplies, dietary supplements, and other regulated items. Recalls are initiated when a product violates FDA laws, poses health risks, or is defective.
Medical Device Safety Alerts: Overview & FDA Process
Medical device safety alerts are critical communications issued when a device poses a risk to patient health. These can range from recalls and corrections to urgent safety notices. Here’s how the FDA and manufacturers handle them:
| Alert Type | Description | Example |
| Class I Recall | Highest risk – may cause serious injury/death. | Faulty defibrillator failing during use. |
| Class II Recall | Moderate risk – may cause temporary/reversible harm. | Software glitch causing incorrect dosing in an insulin pump. |
| Class III Recall | Low risk – unlikely to cause harm. | Minor labeling errors with no health impact. |
| Correction | Fixing a device issue without removing it from market. | Firmware update to correct a malfunction. |
| Removal | Physically removing a device from use. | Hospital returns defective surgical tools. |
| Safety Communication | FDA warning about potential risks. | Alert on cybersecurity vulnerabilities in pacemakers. |
- FDA’s Medical Device Recalls Page Click Here
- MAUDE Database (Adverse event reports). Click Here
- MedSun Program (For high-risk device monitoring) Click Here
FDA Recall Classifications:
The U.S. Food and Drug Administration (FDA) categorizes recalls based on the potential health risks associated with a defective or violative product.
These classifications help determine the urgency of the recall, the level of FDA oversight required, and the necessary corrective actions.
The FDA classifies recalls into three categories: Class I, Class II, and Class III, each reflecting a different level of risk to consumers.
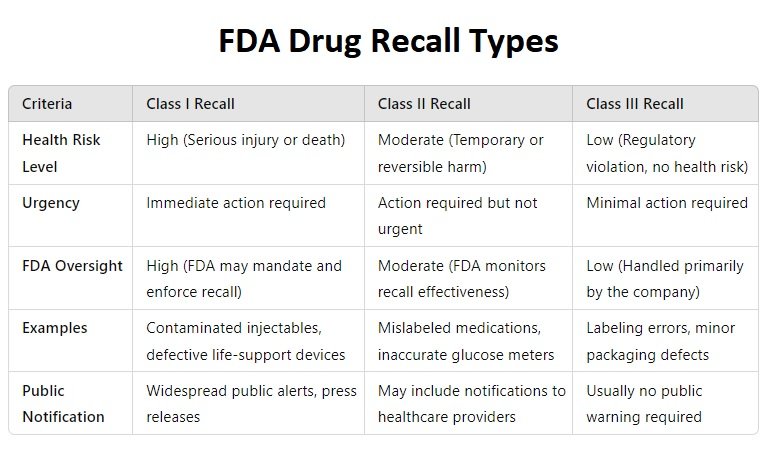
1. Class I Recall (Most Serious)
A Class I recall is issued when a defective product poses a serious threat to health or has the potential to cause death. These recalls require immediate action to prevent harm.Criteria for Class I Recalls
- The product has a high probability of causing serious injury or death.
- There is no acceptable risk level, meaning even a single exposure could be dangerous.
- Immediate removal or correction of the product is necessary to protect consumers.
- The recall may require urgent communication through press releases, public warnings, and direct healthcare provider notifications.
Examples of Class I Recalls
- Defective pacemakers that stop working unexpectedly, leading to cardiac arrest.
- Contaminated injectable drugs containing harmful bacteria that could cause deadly infections.
- Incorrectly labeled high-dose medications, leading to accidental overdose.
- Surgical implants prone to failure, causing severe complications and the need for emergency surgery.
FDA Actions for Class I Recalls
- Immediate and widespread notification to hospitals, healthcare providers, and consumers.
- The FDA may seize products from distribution channels.
- Direct patient and physician communication for urgent medical intervention.
- The FDA ensures the recall is effectively carried out by monitoring consumer response and disposal methods.
Who initiates FDA Class I recalls?
FDA Class I recalls are initiated by either the manufacturer/distributor of the product or by the FDA itself. Here’s how it works:Company-Initiated Recall
- Most recalls (including Class I) are voluntarily initiated by the manufacturer or distributor after they discover a violation or defect that poses a serious risk.
- The company then notifies the FDA, which evaluates and classifies the recall (Class I being the most severe).
FDA-Requested Recall
- If a company fails to act despite identifying a serious hazard, the FDA can formally request a recall.
- In rare cases, the FDA can mandate a recall under specific authorities (e.g., for infant formula, medical devices, or tobacco products).
FDA-Ordered Recall
- For certain products (like defective medical devices or unsafe foods), the FDA has legal authority to order a recall if the company refuses to comply voluntarily.
2. Class II Recall (Moderate Risk)
A Class II recall is issued when a defective product may cause temporary or medically reversible health problems but is not likely to result in serious injury or death.Criteria for Class II Recalls
- The product poses a moderate health risk with the potential for temporary or reversible adverse effects.
- Repeated exposure might increase the likelihood of harm.
- The product remains in circulation for a longer period compared to Class I recalls before corrective actions are fully implemented.
Examples of Class II Recalls
- Mislabeled prescription drugs, where dosage strength is incorrect but within a tolerable range.
- Defective glucose meters providing inaccurate readings that may lead to incorrect insulin dosing.
- Sterility concerns in medical devices like catheters that could lead to infections if used.
- Food supplements containing unapproved ingredients, which may cause minor allergic reactions or digestive issues.
FDA Actions for Class II Recalls
- The FDA monitors the recall process but may not require immediate public notification.
- The manufacturer is typically responsible for contacting distributors and retailers.
- If necessary, the FDA recommends consumer actions (e.g., returning or discarding the product).
- Effectiveness checks may be conducted to ensure defective products are removed from the market.
Class III Recall (Lowest Risk)
A Class III recall is issued when a product violates FDA regulations but is unlikely to cause harm. These recalls involve minor defects that do not pose a significant health threat.Criteria for Class III Recalls
- The defect is not dangerous but still represents a regulatory violation.
- The recall is usually precautionary and done to ensure compliance.
- No documented adverse health effects are associated with the issue.
Examples of Class III Recalls
- Packaging or labeling errors that do not affect product use (e.g., a typo on the label).
- Non-functional components in medical devices that do not impact performance.
- Minor container defects, such as loose bottle caps on over-the-counter drugs.
- Failure to meet regulatory labeling requirements, such as missing expiration dates.
FDA Actions for Class III Recalls
- The recall is typically handled by the manufacturer with minimal FDA involvement.
- The FDA may request product corrections but does not usually require urgent actions.
- Public communication is less common, as these recalls do not pose health risks.
FDA drug Recalls Feed: https://www.fda.gov/drugs/drug-safety-and-availability/drug-recalls
FDA Recall Requirements by Product Category
The U.S. Food and Drug Administration (FDA) oversees recalls for various products, ensuring the safety of medications, medical devices, medical supplies, dietary supplements, and other regulated items. Recalls may be voluntary or FDA-mandated, depending on the severity of the risk. Below is a breakdown of FDA recall requirements by product category.
The Code of Federal Regulations (CFR) is the official compilation of all permanent rules and regulations issued by U.S. federal government agencies. It serves as the detailed implementation of laws passed by Congress and signed by the President.
- Published Annually, updated yearly (with some titles updated quarterly).
- Title 21 (FDA Regulations) – Covers food, drugs, medical devices, cosmetics.
- 21 CFR Part 7 – Recall procedures (including Class I, II, III classifications).
1. Medications (Drugs & Biologics)
Regulated under:- 21 CFR Part 7
What Triggers Recalls for Drugs
- Contamination (bacteria, particulate matter, or foreign substances).
- Incorrect labeling (wrong dosage, missing warnings, or incorrect instructions).
- Stability failures (drug potency degrades before expiration).
- Adulteration (presence of unapproved ingredients or undeclared drugs).
2. Medical Devices
Regulated under:- 21 CFR Part 7
What Triggers Recalls for Medical Devices
- Device malfunctions that could cause harm.
- Sterility failures, leading to infections.
- Software errors affecting device performance.
- Labeling issues that result in improper usage.
3. Medical Supplies (Syringes, Surgical Equipment, Personal Protective Equipment – PPE)
Regulated under:- 21 CFR Part 7 (Recall Guidance)
- 21 CFR Part 820 (Quality System Regulation – QSR)
What Triggers Recalls for Medical Supplies
- Defects affecting function (e.g., faulty syringes that fail to deliver medication).
- Sterility concerns (e.g., surgical gloves contaminated with bacteria).
- Mislabeling (e.g., incorrect size of catheters or gloves).
4. Dietary Supplements
Regulated under:- 21 CFR Part 7 (Recall Guidance)
- Dietary Supplement Health and Education Act (DSHEA)
- 21 CFR Part 111 (Current Good Manufacturing Practices – cGMP)
What Triggers Recalls for Dietary Supplements
- Presence of undeclared drugs or harmful substances.
- False/misleading health claims.
- Contamination with heavy metals or bacteria.
5. Food and Cosmetics
Regulated under:- 21 CFR Part 7 (Recall Guidance)
- Federal Food, Drug, and Cosmetic Act (FD&C Act)
What Triggers Recalls for Food and Cosmetics
- Food contamination (bacteria, undeclared allergens, or foreign objects).
- Cosmetic adulteration (unsafe ingredients or microbial growth).
6. Tobacco and E-Cigarette Products
Regulated under:- 21 CFR Part 7 (Recall Guidance)
- Family Smoking Prevention and Tobacco Control Act
What Triggers Recalls for Tobacco and E0cigarette Products
- E-cigarette devices overheating or exploding.
- Mislabeling of nicotine content.
- Contamination with harmful chemicals.
FDA Recall Databases & Reporting
The FDA maintains public databases for consumers and healthcare providers to track recalls:- FDA Recall Database – Search for active and past recalls by product category.
- MedWatch (FDA Safety Reporting Program) – Consumers and professionals can report safety concerns.
- Enforcement Reports – Weekly updates on FDA recall actions.
How to Report ADRs to the FDA?
1. MedWatch (FDA’s Safety Reporting System)
- Online: www.fda.gov/medwatch
- Phone: 1-800-FDA-1088
- Mail: MedWatch Reporting Forms can be sent to FDA.
2. Vaccine Adverse Event Reporting System (VAERS)
- Managed by: FDA & CDC (Centers for Disease Control and Prevention).
- Purpose: Tracks adverse events related to vaccines to identify potential safety concerns.
- Healthcare providers (required for serious events).
- Vaccine manufacturers (required for all reported events).
- Patients and caregivers (voluntary).
- Allergic reactions (e.g., anaphylaxis).
- Neurological effects (e.g., seizures).
- Fever, pain, or localized swelling.
- Website: vaers.hhs.gov
- Database: Publicly accessible for transparency.
3. FDA Adverse Event Reporting System (FAERS)
- Managed by: FDA.
- Purpose: Tracks adverse events for drugs and biologics (excluding vaccines).
- Healthcare professionals.
- Drug manufacturers (mandatory for serious cases).
- Consumers and patients (voluntary).
- Severe drug reactions (e.g., liver failure from acetaminophen overdose).
- Drug interactions (e.g., serotonin syndrome from antidepressants).
- Unexpected side effects (e.g., blood clotting issues with certain birth control pills).
- Website: fda.gov/faers
- Database: Publicly accessible with regular updates.
VAERS vs FAERS
| Features | VAERS | FAERS |
| Managed by | FDA & CDC | FDA |
| Focus | Vaccines | Drugs & biologics |
| Who Reports? | Healthcare providers, manufacturers, patients | Healthcare providers, manufacturers, patients |
| Mandatory Reporting? | Yes, for serious vaccine reactions | Yes, for serious drug reactions |
| Data Access | Publicly available | Publicly available |
| Example: | COVID-19 vaccine safety monitoring | Drug recalls like Zantac |